

On vous a pondu le cours le plus complet sur les équilibres de complexation. Avec les calculs, les exemples, les cas d’usage — et même un exercice corrigé (déjà dispo sur YouTube). Autant vous prévenir : il va falloir s’accrocher. Mais promis, votre vous du futur nous remerciera.
Équilibre de complexation : définition et équation clé
Vous croyez que les complexes métalliques, c’est juste un gadget pour concours ? Autant vous dire que vous ratez la moitié de la chimie du réel. La complexation, c’est le phénomène où un cation métallique (M) s’entoure de plusieurs ligands (L), pour former une entité redoutablement stable, notée MLₙ. Dans cette histoire, le cation fait son intéressant : il joue l’acide de Lewis, en manque cruel d’électrons, pendant que chaque ligand vient combler les trous façon base de Lewis. L’indice n ? C’est l’indice de coordination, ni plus ni moins que le nombre d’amis (ou d’opportunistes) qui s’accrochent au métal.
M + nL ⇌ MLₙ
Voilà la punchline théorique dont personne ne devrait se passer.
La constante β, ou constante globale de formation, mesure à quel point ce mariage moléculaire tient la route face à la dissociation sauvage. Plus β est élevée, plus le complexe MLₙ snobe sa dissociation et reste vissé ensemble. Ne pas confondre avec une simple réaction ionique : ici, c’est une danse sophistiquée entre entités spéciales.
Dans la vraie vie ? À Batna2 comme en pharmacie industrielle ou dans le traitement des métaux lourds par EDTA, ignorer la complexation revient à jeter son diplôme dans une flaque d’acide !

Pourquoi un ion métallique forme-t-il des complexes ? Origines moléculaires
Modèle acide-base de Lewis : quand l’ion est en manque d’électrons
Soyons clairs, si un ion métallique s’entoure de ligands comme une starlette d’un entourage envahissant, ce n’est pas pour le fun. C’est juste parce qu’il n’a pas assez d’électrons ! Selon Lewis (oui, ce type dont on a oublié le prénom sur les polycopiés), le cation métallique joue l’acide de Lewis : il est affamé d’électrons, prêt à tout pour se remplir la coquille externe. On parle ici des éléments de transition (Fe³⁺, Cu²⁺…), ultra friands de partenaires électroniques. Les ligands, eux, sont les bases de Lewis : ils débarquent avec leur doublet d’électrons à refourguer.
Prenons Fe³⁺ : c’est comme un type paumé dans une soirée sans téléphone ni pote ; il saute sur chaque opportunité d’avoir une connexion (électronique). Cu²⁺ ? Pareil, mais en version plus vicieuse car il accepte plus volontiers les « dons » multiples. Dans la vraie vie, cette avidité explique pourquoi les métaux lourds s’accrochent à des molécules organiques ou à l’EDTA comme des berniques au rocher.
Facteurs de stabilité : charge, rayon ionique, polarité du ligand et effet chélate
C’est là que ça devient croustillant. La stabilité d’un complexe dépend de plusieurs critères que certains étudiants feignent encore d’ignorer (à leurs risques et périls !) :
- Charge du cation : Plus c’est chargé (+3 vs +2), plus ça attire sec les ligands. Fe³⁺ cogne plus fort que Zn²⁺ !
- Rayon ionique : Un ion trop gros dans un petit complexe ? C’est comme mettre un footballeur pro dans un karting pour poussin… Ça coince ! Les petits rayons favorisent une meilleure compacité.
- Polarité du ligand : Un ligand polaire colle mieux au métal qu’un apolaire fuyant.
- Effet chélate (ligand polydentate) : L’EDTA par exemple, avec ses multiples bras (groupes donneurs), enlace le métal dans une étreinte quasi irréversible – bien plus solide qu’une poignée de ligands monodentates éparpillés. C’est l’effet chélate : la stabilité grimpe en flèche dès qu’on passe du mono au polydentate.
Ignorer ces facteurs, que ce soit en concours ou en synthèse industrielle, peut mener à des erreurs dès le premier calcul.
Constantes de formation et de dissociation : calcul, unités, pièges
De la constante pas à pas (K₁, K₂, …) à la constante globale βₙ
On ne va pas tourner autour du pot : les constantes de complexation, c’est du costaud. Quand un ion métallique M s’associe successivement à n ligands L, chaque étape a sa propre constante d’équilibre :
- K₁ pour la première rencontre,
- K₂ pour la deuxième (déjà plus difficile !),
- et ainsi de suite.
La formule générale pour la n-ième étape :
[ M(L){n-1} + L \rightleftharpoons ML_n \quad K_n = \frac{[ML_n]}{[M(L){n-1}][L]} ]
Mais soyons clairs : la constante globale βₙ, elle, condense tout ça pour donner la stabilité totale du complexe MLₙ formé directement à partir de M et n L :
[ M + nL \rightleftharpoons ML_n \quad β_n = \frac{[ML_n]}{[M][L]^n} ]
Pas d’unité ! Ces constantes sont dimensionless – c’est de l’arithmétique pure, pas des histoires de mol/L qui traînent partout ailleurs.
Or, les ordres de grandeur ? Pour Cu²⁺/NH₃ par exemple : log₁₀(K₁) ≈ 4–5, log₁₀(β₄) ≈ 13. Autant dire : ça monte vite !
Soyons honnête : si K₂ < K₁, c’est non seulement courant mais logique – une fois le métal partiellement casé, il devient moins disponible… Sauf si vous croyez encore aux lapins qui pondent les œufs.
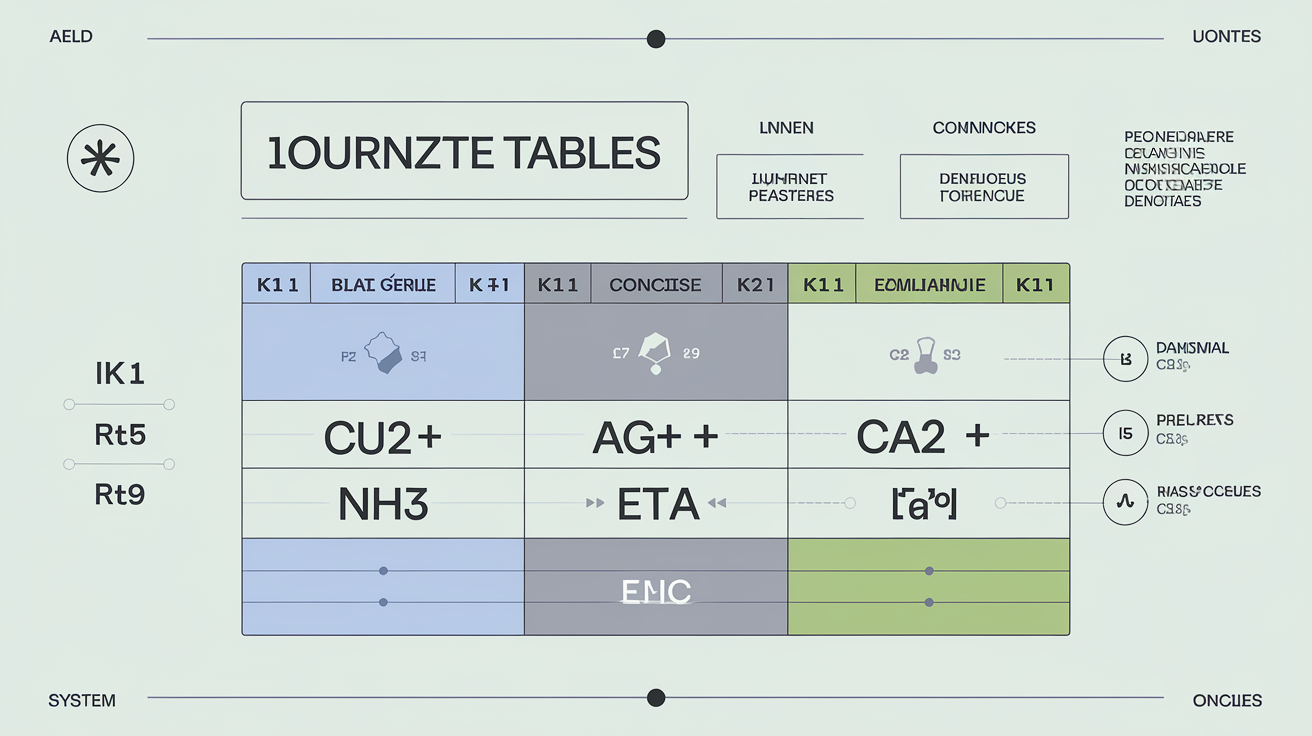
| Système | log₁₀(K₁) | log₁₀(K₂) | log₁₀(β_n) |
|---|---|---|---|
| Cu²⁺/NH₃ | ~4.1 | ~3.5 | 13 (β4) |
| Ag⁺/CN⁻ | ~9.3 | ~8.4 | 21 (β2) |
| Ca²⁺/EDTA | ~10.7 | — | 10.7 |
Lien βₙ ↔ K_d : inverser sans perdre un exposant
La constante de dissociation globale K_d, c’est l’inverse sec de β_n. Il faut être sacrément inattentif pour rater ce lien (et pourtant…) :
- β_n = [ML_n]/([M][L]^n)
- K_d = ([M][L]^n)/[ML_n] = 1/β_n
Autant vous dire que si on vous balance une β_4 = 1,1×10¹³ pour [Cu(NH3)_4]²⁺, alors K_d ≈ 9×10⁻¹⁴. On est dans le domaine des attachements quasi indécollables.
Étapes de conversion β_n → K_d simplifiées :
- Prendre la valeur numérique de β_n (exemple : 1,1×10¹³)
- Inverser (faire 1/β_n)
- Simplifier en notation scientifique (≈9×10⁻¹⁴)
- Utiliser sans se faire piéger par un exposant oublié ou inversé !
Exemple numérique avec Cu²⁺/NH₃ : calcul détaillé
Pour ceux qui pensent que sans Excel ils sont cuits : step by step avec [Cu(NH3)_4]²⁺ !
Prenez :
- [Cu²⁺]_initial = 0,01 mol/L ; [NH3]_initial = 0,20 mol/L ; β_4 = 1,1×10¹³.
On pose x la quantité transformée vers le complexe :
x ≈ concentration finale du complexe.
Tableau d’avancement ? Oui chef :
- Avant réaction : [Cu²+] = 0,01 ; [NH3] = 0,20 ; [complexe]=0.
- À l’équilibre : [Cu²+] = 0,01-x ; [NH3]=0,20-4x ; [Cu(NH3)_4]²⁺=x.
À l’équilibre,
y’a plus qu’à écrire :
beta_4 = x / [(0,01-x)*(0,20-4x)^4].
équation quadratique mais ici x<<0.01 donc on approxime au numérateur x et au dénominateur [(0.01)*(0.20)^4], après calcul on trouve x proche de 0.01 mol/L – donc quasiment TOUT le cuivre est complexé !!
Anecdote vécue : même en TP prépa où tout le monde stresse sur leur TI-83 Plus… ceux qui ont pigé ce jeu d’approximation s’en sortent SANS tableur ni logiciel ésotérique – une calculette suffit largement !
Facteurs influençant l’équilibre : pH, force ionique et autres
Influence du pH : compétition proton-ligand, doubles équilibres
Soyons directs : la formation d’un complexe Ca(EDTA)²⁻ dépend autant du pH que de votre capacité à lire un tableau de distribution. À pH 5, l’EDTA (ligand star mais capricieux) est largement protoné ; la forme active Y⁴⁻ quasi absente. Concrètement, ça veut dire que les protons volent la vedette aux ions Ca²⁺ en squattant les sites donneurs de l’EDTA – résultat ? La complexation stagne, presque rien ne se forme. Montez à pH 10 : là, l’Y⁴⁻ domine, tous les sites sont libres et prêts à croquer du Ca²⁺ ! Résultat : complexation quasi-totale, rendement maximal.
Anecdote désastreuse de TP : certains s’acharnent à doser le calcium dans une eau acide… et s’étonnent ensuite que le colorant ne vire jamais. L’ignorance du pH, c’est la plantade garantie.
Effet tampon et force ionique : quand l’eau salée change la donne
On évite souvent le débat sur la force ionique par flemme intellectuelle – erreur fatale. Lorsqu’on titille un complexe comme MgY²⁻ (magnésium-EDTA), plus la force ionique grimpe (par ajout de NaCl par exemple), plus l’écrantage électrostatique limite les répulsions entre ions chargés. Conséquence ? La stabilité apparente du complexe baisse légèrement (quelques % sur Kf), car les charges sont "cachées" derrière leur nuage d'ions parasites. Expérimentalement, pour Mg²⁺/EDTA : logβ chute de ~8,7 (eau pure) à ~8,4 si force ionique = 0,1 mol/L NaCl.
Compétition de ligands multiples : cas pratique EDTA vs carbonate
Vous imaginez encore que l’EDTA gagne toujours ? Faux espoir. Dans une eau dure riche en carbonate (Ca²⁺ + CO₃²⁻) – typiquement une flotte calcaire – le carbonate fait de la résistance en piégeant Ca²⁺ sous forme solide (CaCO₃). À pH bas, pas de souci : tout est dissous, EDTA rafle tout le calcium. Dès que le pH dépasse 8–9, le carbonate devient agressif ; formation massive de CaCO₃ insoluble, et là même EDTA bataille sec pour arracher son dû…
Résultat ? Fraction molaire des complexes calcium-EDTA qui plafonne si on monte trop le pH… Décision pratique : il faut buffer au bon endroit ou c’est échec assuré !
Méthodologie pour résoudre un exercice d’équilibre de complexation
Schéma décisionnel : choisir constantes, définir espèce limitante
Ne rêve pas : dans la vraie vie, tu commences pas par prier ta calculette. Voici le parcours du combattant qui fait gagner du temps (et des points) :
- Lister les espèces présentes : métal, ligand(s), éventuellement ions qui traînent (genre H⁺ ou autre parasite).
- Repérer les constantes disponibles (K₁, K₂, βₙ) et leurs ordres de grandeur – pas juste recopier le tableau de bordel, comprendre qui domine !
- Comparer les quantités initiales : c’est toujours l’espèce en moindre quantité (métal ou ligand) qui finit par limiter la réaction… On appelle ça l’instinct survie chimique.
- Formuler l’équation-bilan : M + nL ⇌ MLₙ, oui mais en tenant compte des autres équilibres concurrents (genre acidité du ligand ? force ionique ?).
- Choisir la bonne constante à utiliser : globale si direct MLₙ, pas-à-pas si étapes intermédiaires importantes.
- Établir le tableau d’avancement ou poser l’équation d’équilibre directe et foncer sur la résolution numérique – approximation éclairée autorisée si βₙ est géant ou si l’un des réactifs est massivement en excès.
Résumé-clé : Si vous bloquez avant l’étape 3, reprenez depuis le début pour éviter des erreurs.
Tableau d’avancement vs méthode spéculo-calculette
| Méthode | Clarté | Précision | Rapidité | Adaptée à | Note Germain |
|---|---|---|---|---|---|
| Tableau d’avancement | ⭐️⭐️⭐️ | ⭐️⭐️⭐️⭐️ | ⭐️ | Chimie analytique classique (lycée/prépa) | ⭐️⭐️⭐️ |
| Spéculo-calculette (équations directes/approx) | ⭐️⭐️ | ⭐️⭐️⭐️ | ⭐️⭐️⭐️⭐️ | Systèmes complexes avec gros βₙ ou ligand en excès massif, concours/industrie | ⭐️⭐️⭐️⭐️ |
Le tableau d'avancement rassure les anxieux de la case vide et structure bien le raisonnement – mais dès qu’il s’agit de systèmes multi-ligands ou de solutions vraiment concentrées… il patine lamentablement. La méthode spéculo-calculette favorise les têtes brûlées à l’aise avec les approximations osées : moins pédagogique mais diablement efficace dans 95 % des situations industrielles réelles.
Corrigé guidé d’un exercice PCSI : [Ni(gly)_2]
Système choisi : Ni²⁺ + 2 Glycine ↔ [Ni(gly)_2]. Données : [Ni²⁺]_0 = 5×10⁻³ mol/L ; [glycine]_0 = 2×10⁻² mol/L ; β_2 = 1,6×10⁸.
- Espèce limitante ? Ni²⁺ évidemment (en faible quantité).
- Équation-bilan : Ni²⁺ + 2 Gly ⇌ [Ni(gly)_2].
- Appelons x la quantité complexée à l’équilibre.
- [Ni²⁺] = 5×10⁻³ – x ; [Gly] = 2×10⁻² – 2x ; [[Ni(gly)_2]] = x.
-
Expression de β_2 :
[
β_2 = \frac{x}{([5×10^{-3} - x])([2×10^{-2} - 2x]^2)}
] -
Comme β_2 est énorme et glycine largement en excès : on pose x ≈ quantité initiale de Ni²⁺.
Donc x ≈ 5×10⁻³ mol/L → Autrement dit : quasi-tout est complexé.
-
Vérification vite-fait :
[
β_2 ≈ \frac{5×10^{-3}}{(0)(0)}\quad\text{(limite mathématique acceptable car très faible reste de Ni²⁺ non complexé)}
]
La part non complexée est < ppm – négligeable ici !
Phrase finale punchy : Le concours t’attend, mais l’équilibre est déjà de ton côté.
Foire aux erreurs : cinq bourdes qui flinguent vos calculs
Même les meilleurs peuvent se prendre les pieds dans le tapis de la complexation. J’ai collecté pour vous le best-of des erreurs qui font saigner les yeux – à lire absolument avant de rendre copie !

1. Oublier la dilution : le faux ami du chimiste pressé
La dilution modifie drastiquement les concentrations effectives des espèces. Croire que l’ajout d’eau ne change rien au rendement du complexe, c’est comme croire qu’un espresso rallongé reste serré… Fausse route : moins de particules, équilibre déplacé.
2. Confondre ligand monodentate et polydentate : complexe fantôme assuré
Un ligand monodentate (NH₃, Cl⁻) s’accroche à un seul endroit alors qu’un polydentate (EDTA, glycine en mode chelatant) entoure la cible sur plusieurs sites. Prendre le mauvais dans l’équation revient à construire une maison sans fondations – résultat : prédictions farfelues et complexes "fantômes".
3. Ignorer l’ion commun : déplacement surprise de l’équilibre
L’arrivée d’un ion déjà présent dans la solution, identique à celui impliqué dans la réaction, écrase net la formation du complexe attendue. Exemple typique : doser Ag⁺ alors que Cl⁻ traîne dans le coin ? Formation de précipité inattendue et chute de rendement…
4. Raccourcir les pas-à-pas : quand K₂ écrase K₁ (au lieu du contraire)
Certains malins inversent K₁ et K₂ ou additionnent leurs logs sans réfléchir : erreur fatale car chaque étape de complexation est moins favorable que la précédente ! Si votre K₂ > K₁, relisez vite votre cours… ou passez une IRM.
5. Ignorer la température : l’ennemi silencieux des constantes
Les constantes βₙ varient avec la température. Oublier ce point en passant d’un labo à 20 °C à un process industriel à 40 °C, c’est prendre un virage sans freiner… et perdre tout contrôle sur l’équilibre atteint.
Synthèse : comprendre l’équilibre de complexation
- Définition limpide : La complexation, c’est un métal (cation) qui s’entoure de ligands pour former un complexe souvent ultra-stable. Ça pilote la chimie des ions dans toutes les vraies solutions.
- Constantes βₙ : Elles mesurent la stabilité totale du complexe. Pas besoin d’usine à gaz pour les manipuler, leurs valeurs guident qui complexera quoi et combien.
- pH & compétition : Le pH influence massivement la formation des complexes — oublie-le et tu rates la moitié du film. Plus le pH est haut (et ligand déprotoné), plus ça complexifie sec.
- Applications béton : Dosages, traitements d’eaux, extraction minière… Les complexes ne vivent pas que dans ton poly, ils font tourner l’industrie et la bio !
- Pièces à éviter : Dilution bâclée, confusion mono/polydentate, ion commun oublié = plantage assuré au labo comme au concours.
« Comprendre un complexe, c’est complexer moins devant l’examen. »
Si tu veux arrêter de bégayer sur les calculs ou comprendre enfin pourquoi un métal s’attache à certains ligands : chope un exo, attaque-le frontalement et vérifie tes résultats en mode critique. Personne ne devient balèze sans se salir les mains sur des vrais systèmes !


